解锁基因密码之重测序(从测序到分析)
在生命科学的奇妙世界中,基因恰似一本记录着生命奥秘的“天书”,它承载着生物体生长、发育、衰老乃至疾病等一切生命现象的关键信息。而重测序技术,则是开启基因“天书”奥秘的一把神奇钥匙。
试想,你手中有一本经典书籍的通用版本,但在不同的印刷批次中,可能会出现一些细微的错别字、排版差异,甚至独特的批注。基因的情况也是如此,尽管我们已经拥有了某个物种的参考基因组,但每个个体的基因组都可能与参考基因组存在差异。重测序技术的作用就在于此——它通过对已知基因组序列的物种进行个体或群体的基因组测序,然后将测得的序列与参考基因组进行比对,从而精准地找出其中的差异。这些差异或许微小,却可能蕴含着决定个体特征、适应性甚至疾病易感性的关键线索。
测序技术的发展
第一代测序技术
让我们把时光倒回20世纪70年代,第一代测序技术在这个时期应运而生。当时,Sanger和Gilbert这两位科学家分别建立了DNA双脱氧测序和化学测序技术,为基因测序领域奠定了坚实的基础。
先来看DNA双脱氧测序法,其核心原理是利用双脱氧核苷酸(ddNTP)来终止DNA链的延伸。在反应过程中,当ddNTP随机掺入到正在合成的DNA链中时,由于它缺乏继续延伸所需的化学基团,链的生长就此停止。这样,就会产生一系列不同长度的DNA片段。随后,通过电泳技术将这些片段按照大小分离,再利用放射自显影来阅读DNA序列。
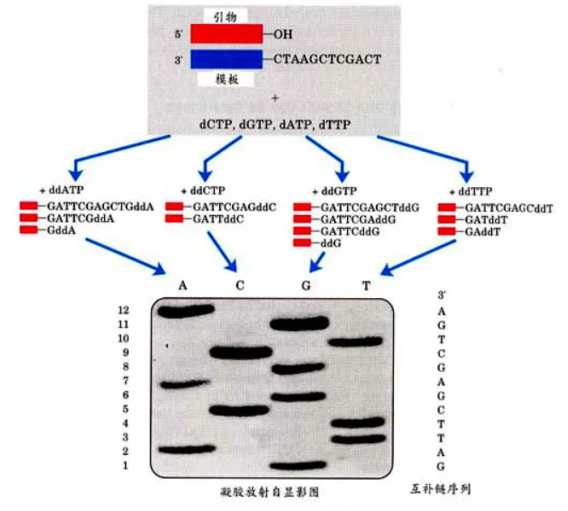
图1 DNA双脱氧测序法
而化学测序法的原理则有所不同,它是通过特定的化学反应,使DNA链在不同的碱基位置处发生断裂。同样地,经过电泳分离和放射自显影处理后,便能够确定DNA的序列。
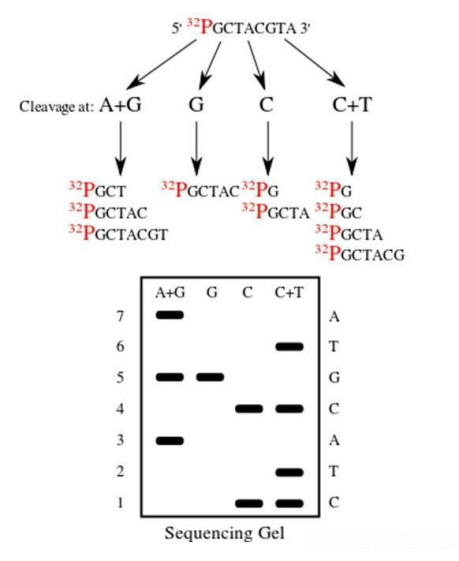
图2 化学测序法
第一代测序技术的特点十分鲜明,它的准确性非常高,这是其最大的优势所在。然而,该技术也存在一些明显的不足:a)通量较低,无法同时处理大量的样本;b)成本相对较高,对于大规模的基因组测序项目来说经济负担较重;c)操作过程较为繁琐,需要耗费大量的人力和时间;d)测序速度缓慢,难以满足快速获取基因信息的需求。一次测序只能读取几百个碱基对的序列信息,这在面对庞大的基因组时显得力不从心。尽管如此,第一代测序技术在当时依然为生命科学的研究提供了重要的工具,推动了遗传学等领域的发展。
第二代测序技术(高通量测序技术)
时间的车轮滚滚向前,来到了20世纪末至21世纪初,这一时期见证了第二代测序技术,也就是我们常说的高通量测序技术的崛起。
在技术层面,这一代测序技术呈现出了百花齐放的局面。罗氏454的焦磷酸测序技术率先登场,它巧妙地利用了焦磷酸的释放与检测来推断碱基的掺入,从而实现测序。紧随其后,Solexa的SBS测序技术(后续被Illumina公司收购,并发展成为如今广为人知的HiSeq等系列测序平台)也崭露头角,该技术通过边合成边测序的方式,在每一次碱基掺入时进行荧光信号检测,进而读取DNA序列。此外,Complete Genomics(CG)的DNB连接cPAL测序技术也加入了这一技术浪潮,最终被华大基因收购并发展成为华大智造的一系列BGI/MGI测序平台。还有 SOLiD 测序技术等,它们共同构成了第二代测序技术的多元格局。目前爱基测序平台有Illumina和华大MGI。
从原理上来说,这些技术有着共通之处,它们都是基于DNA合成或连接反应来进行测序。在具体操作中,首先将基因组DNA或RNA打断成小片段,然后连接上特定的接头,就像是给每个片段都装上了“把手”,方便后续的操作。接着,这些带有接头的片段被放置在固相表面上,在这里,无数个测序反应可以同时进行,也就是我们所说的多重平行测序反应。在测序过程中,每一个循环的数据都会通过成像的方法被捕捉下来,随着循环的不断进行,序列信息也就被逐步解析出来,从而实现了大规模、高通量的测序。
第二代测序技术的特点十分突出,最令人瞩目的是测序通量得到了大幅提升,相较于第一代测序技术,这是一次质的飞跃。同时,成本也显著降低,使得大规模基因组测序项目在经济上变得可行。它能够同时对大量DNA片段进行测序,一次运行可以产生数百万到数十亿条序列信息,这为生命科学研究开辟了广阔的道路。然而,任何技术都不是完美的,第二代测序技术的缺点在于测序读长短,这在一定程度上限制了对某些复杂基因组区域的精确分析。不过,瑕不掩瑜,第二代测序技术的出现,使得大规模基因组测序成为可能,极大地推动了生命科学各个领域的发展,从基础研究到临床应用,都留下了它深深的印记。
图3 Illumina扩增原理
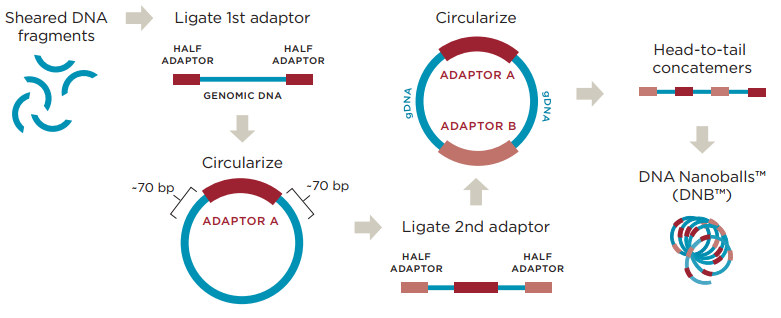
图4 华大扩增原理
第三代测序技术
21世纪以来,随着生命科学的不断探索与技术的飞速发展,第三代测序技术崭新登场,它犹如一颗璀璨的新星,照亮了基因测序领域的前行道路。
在技术层面,第三代测序技术以单分子测序技术为核心,其中最具代表性的当属PacBio RS测序技术和纳米孔测序技术。PacBio RS测序技术就像是一个敏锐的实时监控器,它基于单分子实时(SMRT)测序原理,在DNA聚合酶催化DNA合成的过程中,通过荧光标记的核苷酸发出的荧光信号,实时捕捉并监测DNA序列的合成过程,仿佛亲眼目睹每一个碱基的精准掺入。
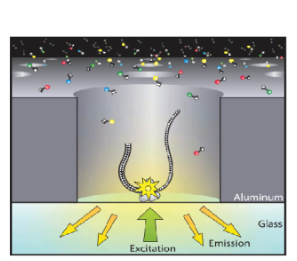
图5 PacBio平台测序
而纳米孔测序技术则犹如一个神奇的电流探测器,它让DNA分子如同穿针引线般通过纳米孔,当不同的碱基依次通过这个微小的孔道时,会引起纳米孔电流的微妙变化,就像不同的钥匙插入锁孔产生的不同震动,通过敏锐地检测这些电流变化,就能准确地确定DNA序列。
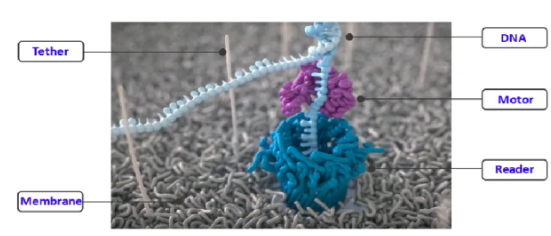
图6 Nanopore平台测序
第三代测序技术的特点更是令人眼前一亮。它能够实现单分子水平的测序,无需再借助PCR扩增这一传统步骤,这就如同直接从源头获取信息,避免了扩增过程中可能引入的错误和偏差,保证了原始DNA分子的完整性与真实性。同时,它还具备检测特殊碱基修饰等信息的能力,为研究基因的表观遗传调控等复杂机制提供了有力支持。
此外,其测序读长较长,就像是拥有了一把更长的尺子,能够轻松跨越一些复杂的基因组区域,例如那些重复序列、高变异区域等,这极大地有助于基因组的精细组装和结构变异的精准检测,为研究人员呈现出更加完整、清晰的基因组画卷。
然而,如同每一枚硬币都有两面,第三代测序技术目前也存在一些亟待解决的问题。测序成本相对较高,这在一定程度上限制了其大规模应用的范围,就好比一件精美的手工艺术品,由于制作成本高昂,难以普及到大众手中。同时,其准确性还有待进一步提高,虽然已经能够满足许多研究需求,但在追求极致精确的科学道路上,仍有提升的空间。
重测序技术产品
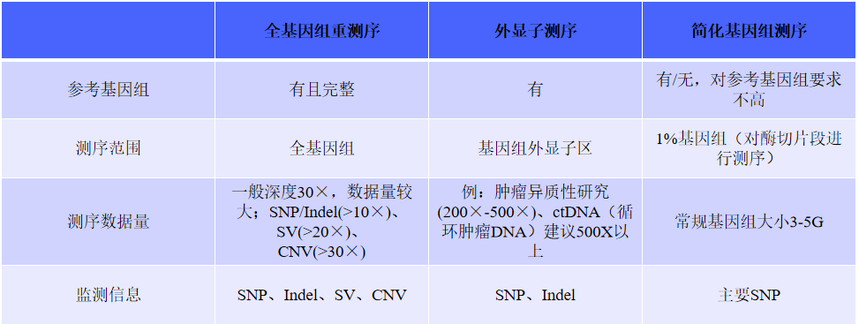
一、全基因组重测序——基因全景大扫描
全基因组重测序(Whole - Genome Resequencing,WGR)相当于对生物体基因组进行一次全面的“体检”,它的视野覆盖了基因组的每一个角落,无论是负责编码蛋白质的关键区域,还是曾被误解为“垃圾DNA”的部分,都在它的检测范围内。这就好比用高清摄像机对整个城市进行全方位、无死角的拍摄,不遗漏任何一个细节。
通过全基因组重测序,我们可以精准地检测出基因组中的各种变异类型。其中,单核苷酸多态性(SNP)就像是基因组中的“单个音符变化”,虽然微小,却可能影响生物性状;插入缺失(InDel)则像是“音符的增减”,对基因序列的长度产生影响;而结构变异(SV)和拷贝数变异(CNV)更是涉及基因组的“乐章重组”,可能导致基因功能的改变、基因表达的失调等复杂后果。
在个体水平上,全基因组重测序能够实现基因型多样性分析,揭示个体独特的基因组成;进行遗传进化分析,追溯个体的遗传起源与演化历程;筛选致病和易感性基因,为疾病的风险评估提供依据;开展单基因病筛查,精准定位由单个基因突变引起的疾病;以及实施癌症筛查,发现与癌症发生发展相关的基因变异,为早期诊断和治疗提供可能。
在群体水平上,通过对多个个体的全基因组重测序,可以挖掘出群体内丰富的遗传变异,为研究群体的遗传结构、基因流动、自然选择等提供海量数据,有助于我们深入了解生物的进化历史、适应性机制以及疾病的群体遗传学特征,为生命科学的众多领域带来新的突破和应用场景。
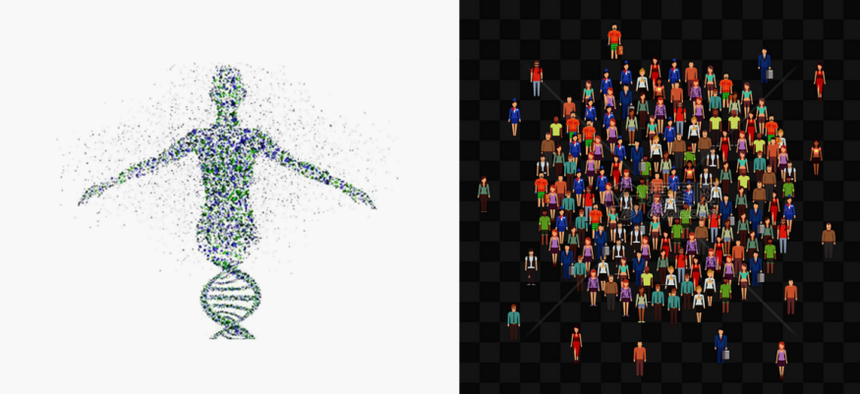
图7 个体与群体
二、外显子测序——聚焦关键“剧情”
在基因组这部浩瀚的“生命史诗”中,全外显子组测序(Whole Exon Sequencing,WES)就像是一位专注的“剧情推动者”追踪者,它专注于那些真正决定生物性状和功能的关键章节——外显子。外显子是基因中能够编码蛋白质的区域,它们虽然只占基因组的1%,但却承载着大约85%的致病突变,是疾病相关变异的“高发区”。
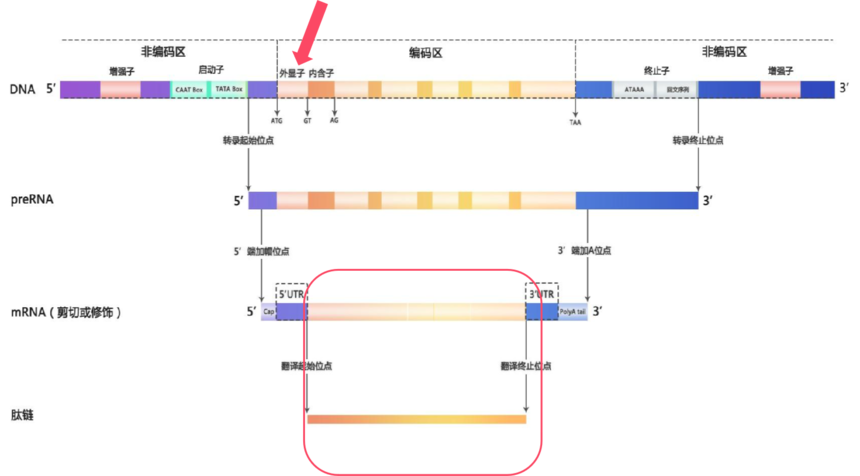
图8 真核生物基因结构
WES借助序列捕获技术,如同精准的“基因渔网”,将全基因组中外显子区域的DNA捕获并富集,随后进行高通量测序。这一过程就像是直接对外显子这些关键章节进行精细的“文字校对”,能够更准确地发现与蛋白质功能变异相关的遗传变异。
与全基因组重测序相比,外显子测序由于数据量较小,使得成本大幅降低,这就好比只精读关键部分而不是整本书,节省了大量时间和资源。同时,其在有效位点的测序深度却更高,意味着对这些关键区域的检测更加精细和准确。
在医学研究领域,WES堪称“疾病基因猎手”,常用于查找单基因病、复杂疾病的致病基因,以及检测肿瘤的体细胞突变。它是揭示疾病发生机制、寻找治疗靶点的重要工具。
WES在肿瘤研究方面具有独特优势。癌症异质性需要高深度测序来全面揭示其复杂性,建议达到200X以上有效深度,以确保对肿瘤基因组的精准解析。对于FFPE(甲醛固定石蜡包埋)样品,由于其在处理过程中可能造成DNA损伤,建议采用200 - 300X对应的数据量,以保证数据的可靠性和准确性。而在检测ctDNA(循环肿瘤DNA)时,为了捕捉到血液中微量的肿瘤DNA信号,建议达到500X及以上有效测序深度。这些精细的测序策略有助于我们深入理解肿瘤的发生、发展、转移和复发机制,以及评估药物疗效,为肿瘤的精准治疗和个性化医疗提供有力支持。
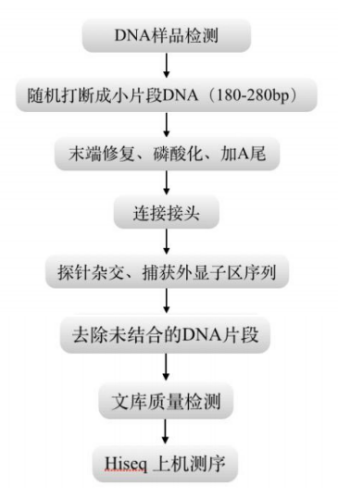
图9 WES测序流程
三、简化基因组测序 —— 高效抓重点
在基因组这片广袤的“信息海洋”中,简化基因组测序技术(Reduced Representation Genome Sequencing,RRGS)就像是一艘精准的“信息采集船”,它包括RRL、GBS、RAD-seq、ddRAD和2b-RAD等多种方法,通过限制性内切酶消化DNA来降低基因组复杂度,再利用测序技术进行遗传变异分析。这一过程相当于从海量信息中精心挑选出最具代表性的部分进行测序,就像是在繁杂的图书馆中挑选出关键的书籍来阅读,从而有效减少了测序的数据量,避免了不必要的信息干扰。
RRGS技术凭借其成本低、效率高的优势,成为了大规模样本测序的“利器”,能在短时间内对大量样本进行测序分析,这就好比一条高效的生产线,快速而稳定地输出精准的数据。在群体遗传学研究中,它更是发挥了巨大的作用,帮助我们深入地了解群体的遗传结构和多样性,揭示不同群体之间的演化关系和遗传差异,为生物进化研究和育种工作提供了宝贵的数据支持。
-
简化基因组测序手段的各自特点:
· RRL(Reduced Representation Libraries):
RRL操作相对简单,能够快速构建文库,就像是一个“快速启动的项目”,可以迅速投入运行并产生数据。但可能产生较多的非特异性片段,导致测序数据中有效信息占比相对较低,这就好比在采集的样本中夹杂了一些杂质,需要进一步的筛选和纯化。此外,它对酶切位点的依赖性较强,酶切位点的分布会影响所获得的基因组覆盖范围,这意味着在实验设计时需要充分考虑酶切位点的选择和分布,以确保获得具有代表性的基因组区域。
· RAD-seq(Restriction-site Associated DNA sequencing):
RAD-seq依赖机械剪切和酶切结合的方式,这种独特的组合使其适用于缺乏参考基因组的物种,就像是为那些“神秘的、未被充分研究”的生物量身定制的工具,能够在没有完整基因组信息的情况下,依然精准地获取关键的遗传信息,为研究这些物种的遗传多样性和适应性进化提供了可能。
· GBS(Genotyping By Sequencing):
GBS可以看作是一种简化版的复杂度降低方法,它仅使用一种限制性酶,从而大大简化了操作流程和成本,就像是一个“经济适用型”的测序方案,适用于多种物种的低成本基因分型,在大规模的群体筛查和遗传标记辅助育种中具有广泛的应用前景,能够快速、高效地为育种工作提供大量的遗传标记数据。
· ddRAD(double Digest Restriction-site Associated DNA):
ddRAD使用两种限制性酶进行消化,这种双酶切策略提供了更高的位点控制,就像是给基因组信息的采集安装了“双保险”,使得获取的遗传信息更加精准和全面。它适用于遗传连锁图谱构建和关联分析,在研究基因的连锁关系、定位重要农艺性状的基因位点等方面具有独特的优势,为作物和家畜的分子育种提供了有力的工具。
· 2b-RAD:
2b-RAD基于IIB型限制性内切酶,这种特殊的酶切方式能够生成等长标签,就像是生产出了一批标准化的“遗传信息标签”,便于统一分析和比较,适用于大规模研究和高密度遗传图谱构建。在需要处理大量样本和构建精细遗传图谱的研究中,2b-RAD能够高效地完成任务,为揭示复杂性状的遗传基础和开展精准育种提供了重要的技术支撑。
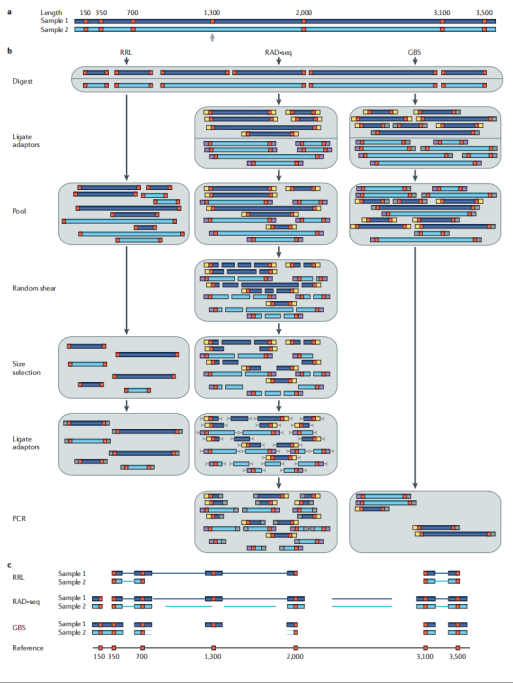
图10 简化基因组测序原理
重测序数据分析应用
一、群体进化研究
在生命科学的探索中,群体进化研究宛如一把开启物种历史奥秘的钥匙,它基于全基因组重测序技术或其他测序技术,为我们揭示生物群体的遗传奥秘。通过获取某物种自然群体各亚群的基因组信息,我们能够得到大量的SNP、InDel、SV和CNV等变异信息,进而深入研究群体的遗传结构、驯化机制、种群历史以及群体进化动态等生物学问题,勾勒出物种演化的壮丽画卷。
-
应用方向
物种起源与演化探究
通过分析不同物种或群体的重测序数据,我们可以重建物种的进化历程,确定物种的起源时间和地点,以及不同物种之间的亲缘关系。这就像为物种的家族树绘制详细的年轮,每一道刻痕都记录着时间的流逝和生命的变迁。
基因功能与调控网络研究
群体进化研究可以结合重测序数据和表达谱数据,推断基因的功能和调控网络。在不同环境或发育阶段下,群体中基因的变异和表达变化模式能够为理解基因的功能及其在生物体内的调控机制提供线索,为我们揭示生命活动的内在逻辑。
农业种植领域可进行作物遗传改良
在作物育种中,重测序数据可用于挖掘与重要农艺性状相关的基因和标记。例如,通过对大量水稻品种进行重测序,发现与产量、品质、抗病性等性状相关的基因位点,为分子标记辅助选择育种和基因编辑育种提供理论依据,助力农业的丰收与进步。
家畜品种选育
对于家畜,重测序数据有助于了解其遗传多样性和群体结构,鉴定与生长速度、肉质、繁殖性能等经济性状相关的基因。通过对猪、牛等家畜群体的重测序研究,能够有针对性地进行品种选育,提高养殖效益,为畜牧业的发展添砖加瓦。
医学领域可进行疾病遗传机制研究
对患者群体和健康群体进行全基因组重测序,对比分析两者之间的遗传差异,发现与疾病发生发展相关的基因突变和易感位点,为疾病的诊断、治疗和预防提供重要的科学依据,守护人类的健康。
-
材料选择
种群需具有长时间的进化或驯化历史,可选取全球范围内不同地区、不同品种样本研究该种群的动态历史;或者选取某特定地区不同品种样本,研究该种群在该地区的种群历史。
-
样本数量
至少3个群体,建议每个分群植物不少于15个,动物不少于10个,珍稀样品数量可适当减少。
-
测序深度
一般建议10×以上,珍稀样本可适当减少。
-
分析内容展示
① PCA(主成分分析)
通过数据降维分析主成分,揭示数据中的主要变异模式和样本间的关系。在群体进化研究中,用于分析群体遗传结构,直观地展示不同群体间的遗传差异和相似性,判断群体是否存在明显的遗传分化,以及个体在群体中的遗传位置等。
② 进化树
基于生物之间的遗传距离或相似性,通过一定的算法构建树形结构,以表示不同生物类群或个体之间的进化关系。树的节点代表祖先群体或物种,分支长度通常反映了进化距离或遗传差异的大小。
③ Structure 分析
基于群体遗传学的模型,利用个体的基因型数据,将群体中的个体按照遗传相似性划分成不同的遗传簇;用于研究群体的遗传混合、基因流和种群分化情况,确定群体中存在的潜在亚群数量和结构,分析个体在不同亚群中的遗传组成比例。
④ 选择性清除分析
当一个有利突变在群体中受到自然选择作用而迅速传播时,它会导致该突变位点及其附近的基因组区域的遗传多样性降低,这种现象称为选择性清除。通过比较不同群体或同一群体不同区域的遗传多样性、等位基因频率等指标,来识别基因组中可能受到正选择的区域。
⑤ π 分析
π 值是衡量群体内遗传多样性的一个指标,它表示群体中任意两个随机选择的 DNA 序列之间核苷酸差异的平均数。通过计算群体中基因或基因组区域的 π 值,了解该群体在特定区域的遗传多样性水平。评估群体的遗传变异程度,比较不同群体之间或同一群体不同区域的遗传多样性高低,分析遗传多样性在群体中的分布情况。
⑥ Fst 分析
是衡量群体间遗传分化程度的一个统计量,反映了群体间基因频率的差异程度。基于群体遗传学的 Hardy-Weinberg 平衡原理,通过比较群体内和群体间的基因多样性来计算。Fst 值的范围在 0 到 1 之间,值越大表示群体间的遗传分化程度越高。
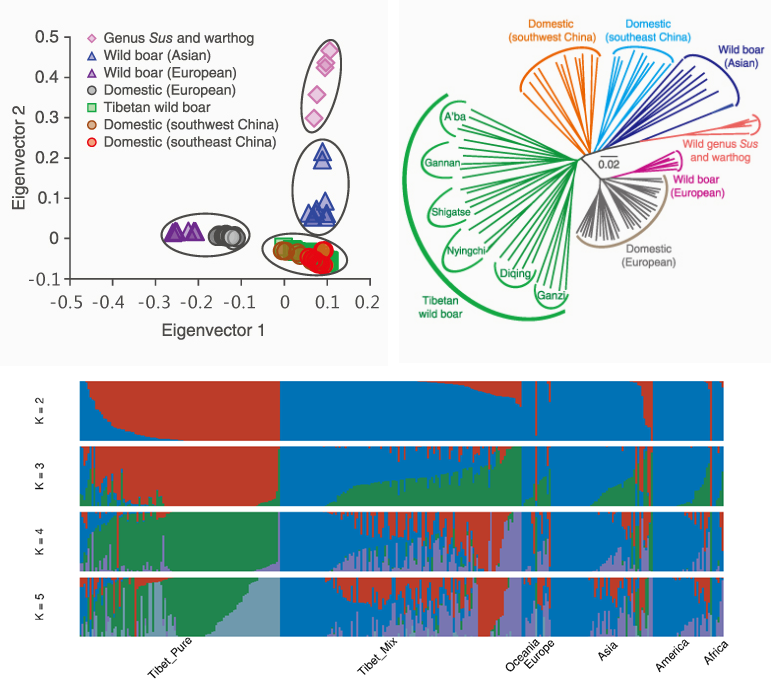
图11 PCA、进化树与structure分析
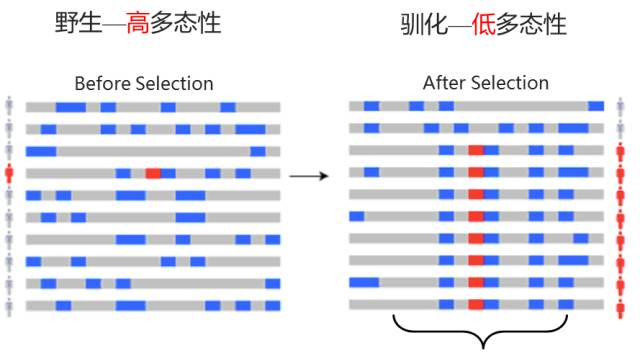
图12 选择性清除分析
二、全基因组关联分析GWAS
全基因组关联分析(Genome wide association study,GWAS)是一种在全基因组范围内对多个个体的遗传变异(标记)多态性进行检测,以获得基因型数据,并将这些基因型与可观测的性状(表型)在群体水平上进行统计学分析的方法。通过分析统计量或显著性 p 值,GWAS 能够筛选出最有可能影响特定性状的遗传变异(标记),进而挖掘出与性状变异相关的基因,为揭示复杂性状的遗传基础提供有力支持。
-
数据要求
表型数据
为了确保分析结果的可靠性和准确性,表型数据的收集应尽可能全面和精确。通常情况下,建议在多年份、多地点的条件下进行数据采集,并且每个处理或条件下的重复次数最好达到三次以上。这种设计有助于充分考虑环境因素对表型性状的影响,降低随机误差对实验结果的干扰,从而更真实地反映基因与性状之间的关联关系。
测序样本数量
样本数量的多少直接影响到 GWAS 的统计效力和结果的可信度。一般要求样本数大于 200,这样可以确保在统计分析中具有足够的功率来检测到微弱的基因效应,同时也有助于提高所发现关联信号的稳定性。此外,对于自然群体,应尽量选取来自各个地方的不同材料,包括野生型、栽培种以及地方品种等,以涵盖丰富的遗传多样性,增强研究结果的普遍性和代表性。在家系群体的研究中,则可选择全同胞家系或半同胞家系,以及多个亲本及其 F2 子代等作为研究对象,这样能够充分利用家系内的遗传连锁信息,提高定位基因的精度。
测序深度
测序深度与数据质量和分析准确性密切相关。一般建议测序深度达到 10× 以上,这样可以确保在大多数基因组区域获得可靠的测序数据,减少因测序深度不足导致的基因型误判。同时,适当的测序深度也有助于平衡研究成本和数据质量,使得在大规模样本测序的情况下,既能保证数据的可用性,又不会造成资源的过度浪费。
-
分析内容展示
曼哈顿图
曼哈顿图是一种直观展示 GWAS 结果的散点图,因形似曼哈顿摩天大楼而得名。在图中,横轴表示基因组的染色体位置,按照染色体的顺序依次排列,不同染色体通常用不同的颜色加以区分;纵轴则表示 -log10(P),P 值越小,对应的纵轴值越高,表明该位置的遗传变异与目标性状的关联越显著。研究人员可以通过观察曼哈顿图,快速锁定基因组中具有显著关联信号的区域,这些区域中的变异位点即为可能影响性状的关键候选位点,为进一步的功能验证和基因挖掘提供了明确的方向。在实际研究中,尤其是对于数量性状的分析,如果曼哈顿图中出现成簇的显著性位点,往往更具生物学意义,因为多个相邻的显著位点可能共同作用于性状的形成和变异,而单一位点的显著性可能存在假阳性风险,需要谨慎对待。
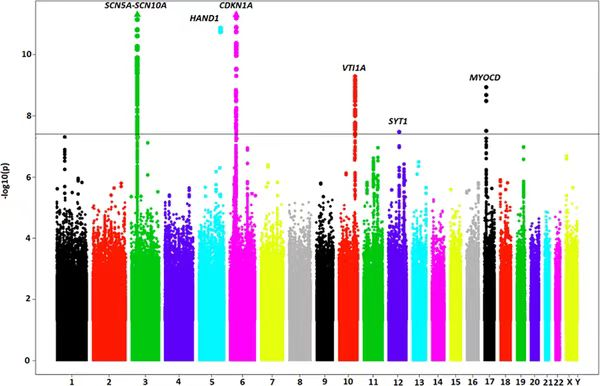
图13 曼哈顿图
QQ 图
QQ 图(quantile-quantile plot),即分位图,是评估 GWAS 分析结果质量的重要工具,主要用于判断观察到的 P 值分布与期望的 P 值分布之间的差异,从而帮助研究人员识别潜在的假阳性或假阴性结果。在 QQ 图中,若观察到的 P 值与期望 P 值一致,数据点将沿对角线(y=x)分布,这表明分析过程中没有明显的系统性偏差,所有的显著性结果均可认为是随机产生的。然而,当数据点偏离对角线时,存在两种情况:一种是向上偏离,即数据点位于对角线之上,这表示观察到的 P 值比期望的更小,意味着存在真实的显著关联信号,可能是由基因与性状之间的实际关联所导致;另一种是向下偏离,数据点位于对角线之下,这可能暗示着存在系统性偏差,例如群体结构未得到充分校正、实验操作中的技术误差等,这些因素可能会干扰分析结果的准确性,导致真正的关联信号被掩盖或弱化。因此,通过仔细分析 QQ 图的形状和特征,研究人员可以对 GWAS 结果的可靠性和有效性进行更为全面和深入的评估,为后续的结论和解释提供更为坚实的依据。
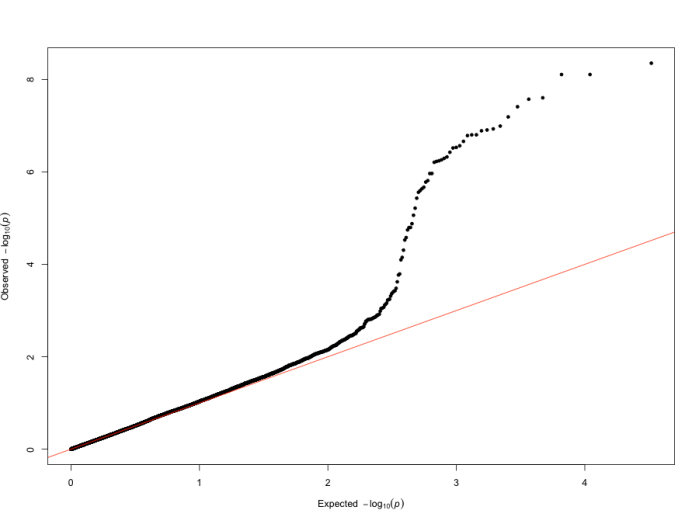
图14 QQ图
注:更加详细的GWAS介绍点击:https://mp.weixin.qq.com/s/ALd4ZxOymKKRbqPItFrRTQ
三、遗传图谱构建和QTL定位
-
遗传图谱
遗传图谱(Genetic map),也称为遗传连锁图(Genetic linkage map),是通过分析遗传标记之间的连锁关系而建立起来的染色体图谱。遗传距离通常用厘摩(cM)来表示,它反映了基因或标记之间在减数分裂过程中发生交换和重组的概率。在植物遗传学研究中,遗传图谱帮助揭示基因在染色体上的相对位置和遗传连锁关系。
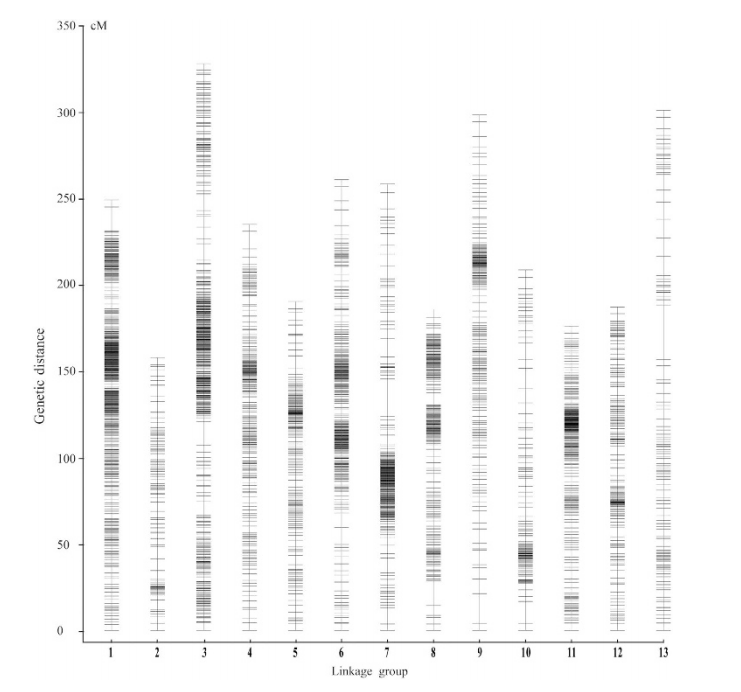
图15 SNP图谱的13个连锁群中的分布[1]
测序样本
选择亲本和构建分离群体:选择具有明显性状差异且纯合的亲本进行杂交,获得 F1 代。然后让 F1 代自交或与亲本回交等,构建 F2 代、回交群体(BC)等分离群体。这些群体中的个体在基因组合上存在差异,是遗传图谱构建的基础材料。

测序深度
建议最少10×以上。对于大多数物种,10x-20x的深度结合合适的统计方法和参考panel等能获得较好的效果来推断群体的遗传特征等;对于具有复杂结构的基因组(如杂合度高的群体或多倍体生物),可能需要更高的测序深度,30x甚至更高,以确保准确捕捉到所有变异。
-
QTL定位
数量性状位点(Quantitative Trait Locus,QTL),是指控制数量性状的基因在基因组中的位置。QTL定位是根据分子标记与目标性状的连锁关系,通过统计方法来定位与目标性状相关染色体区域,这些区域可能包含导致目标性状表型变异的基因。广泛应用于基因组研究、品种改良和基因定位等领域。

图16 QTL准备及分析流程
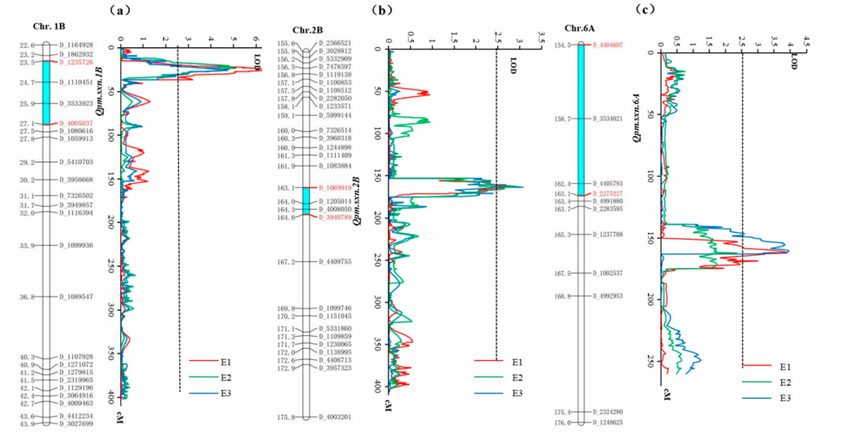
图17 1B(a)、2B(b)和 6A(c)号染色体上白粉病(PM)的数量性状基因座(QTL)及遗传连锁图谱[2]
基于连锁关系的QTL定位
QTL定位到的区间往往包含较多候选基因,可以采用以下辅助研究方法来筛选和验证区间内的候选基因:
-
构建次级分离群体:通过进一步构建更小的分离群体,可以更精细地定位QTL区间,缩小候选基因的范围。
-
结合公共数据库查看功能注释信息:利用已有的基因功能注释数据库,对QTL区间内的基因进行功能分析,筛选出可能与目标性状相关的基因。
-
同源基因分析:通过比较不同物种中的同源基因,寻找可能的功能保守基因,这些基因可能在目标性状的形成中起关键作用。
-
转录组分析:通过对目标性状相关组织或时期的转录组数据进行分析,了解QTL区间内基因的表达模式,筛选出可能与性状变异相关的差异表达基因。
-
基因编辑:利用基因编辑技术对候选基因进行功能验证,通过敲除或编辑基因,观察对目标性状的影响,从而确定基因的功能。
针对高通量测序,爱基百客拥有全基因重测序、全外显子测序、宏基因组和RAD等产品,欢迎有相关研究需求的老师前来咨询~
-
参考文献
【1】Zhang, H., Miao, H., Li, C., Wei, L., Duan, Y., Ma, Q., Kong, J., Xu, F., & Chang, S. (2016). Ultra-dense SNP genetic map construction and identification of SiDt gene controlling the determinate growth habit in Sesamum indicum L. Scientific reports, 6, 31556. https://doi.org/10.1038/srep31556
【2】Zhao, Z., Qiu, Y., Cao, M., Bi, H., Si, G., & Meng, X. (2024). Quantitative Trait Loci Mapping for Powdery Mildew Resistance in Wheat Genetic Population. Genes, 15(11), 1438. https://doi.org/10.3390/genes15111438
相关文章:
)
解锁基因密码之重测序(从测序到分析)
在生命科学的奇妙世界中,基因恰似一本记录着生命奥秘的“天书”,它承载着生物体生长、发育、衰老乃至疾病等一切生命现象的关键信息。而重测序技术,则是开启基因“天书”奥秘的一把神奇钥匙。 试想,你手中有一本经典书籍的通用版…...

当使用 Docker Desktop 启动 Tomcat 镜像时时间不对
当使用 Docker Desktop 启动 Tomcat 镜像时时间不对,可能由以下原因导致并可采取相应解决方法: 宿主机时间设置问题:Docker 容器的时间是由宿主机提供的,如果宿主机的时间不正确,那么容器的时间也会不正确。需确保宿主…...

golang gmp模型分析
思维导图: 1. 发展过程 思维导图: 在单机时代是没有多线程、多进程、协程这些概念的。早期的操作系统都是顺序执行 单进程的缺点有: 单一执行流程、计算机只能一个任务一个任务进行处理进程阻塞所带来的CPU时间的浪费 处于对CPU资源的利用&…...
的区别)
Redisson的RedLock与联锁(MultiLock)的区别
Redisson提供了两种分布式锁机制:RedLock(红锁)和MultiLock(联锁),它们在实现分布式锁时有重要区别。 1. RedLock (红锁) 设计原理: 基于Redis官方提出的Redlock算法实现目的是在Redis集群环境下提供更可靠的分布式锁需要至少3个独立的Redi…...

图灵逆向——题十-魔改算法
目录列表 过程分析JS代码还原代码实现运行结果 本题属于魔改标准加密算法,所以无法使用JS或Python中的标准库来进行模拟加密了,只能一步一步的还原它的环境咯。。。 过程分析 打开控制台发现有个无限debugger,直接过掉~[doge]。。。 OK过掉…...

K8S学习之基础七十九:关闭istio功能
关闭istio功能 kubectl get ns --show-labels kubectl label ns default istio-injection-有istio-injectionenabled的命名空间,pod都会开启istio功能 反之,如果要开启istio,在对应命名空间打上该标签即可...

AI大模型课程系列汇总-某客时间篇
某客时间系列篇章💥 NO.文章(点击可跳转)1【课程系列11】某客时间AI 大模型应用开发实战营_ai大模型应用开发实战营 百度云-CSDN博客2【课程系列12】某客时间AI大模型微调训练营_极客时间 大模型rag 网盘-CSDN博客3【课程系列15】2024某客时…...

【人工智能】大语言模型多义词解析技术揭秘——以“项目“歧义消解为例
今天田辛老师和小伙伴探讨了一个有趣的多义词问题, 在人工智能技术日新月异的今天,大语言模型(LLM)对自然语言的理解能力已经达到令人惊叹的水平。大模型到底是如何去区分多义词的? 比如:当用户提到"…...

Coze平台 发布AI测试Agent的完整实现方案
以下是基于 Coze平台 发布AI测试Agent的完整实现方案,包含架构设计、核心功能模块、集成方式和落地步骤: 一、方案架构图 #mermaid-svg-kzS7cltxrbetjROl {font-family:"trebuchet ms",verdana,arial,sans-serif;font-size:16px;fill:#333;}#mermaid-svg-kzS7cltxr…...
)
OpenHarmony子系统开发 - 调测工具(二)
OpenHarmony子系统开发 - 调测工具(二) 三、hiperf使用指导 hiperf是为开发人员提供性能采样分析的工具,基于内核perf机制进行的用户态能力的扩展,可以对指定的程序或者整个系统进行性能采样。 hiperf支持的命令有:l…...

keil ERROR: L6220E 如何解决
最近调试MK8000方案,增加code后遇到: keil ERROR: L6220E: Execution region RW_RAM1 size (24592 bytes) execeeds limit (24568 bytes) .region contains 93 bytes of padding and 0 bytes of veneers (total 93 bytes of linker generated content) …...

c++比较器——priority_queue用 ; unordered_map 自定义哈希函数
文章目录 priority_queue自定义比较方法对 比较对象结构体 重载 <仿函数为什么是传一个类std::less<T> 和 std::greater<T> lambda unordered_map自定义哈希函数仿函数lambda priority_queue template<class T,class Container std::vector<T>,class…...

centos-LLM-生物信息-BioGPT安装
参考: GitHub - microsoft/BioGPT https://github.com/microsoft/BioGPT BioGPT:用于生物医学文本生成和挖掘的生成式预训练转换器 |生物信息学简报 |牛津学术 — BioGPT: generative pre-trained transformer for biomedical text generation and mini…...

esp32cam远程图传:AI Thinker ESP32-CAM -》 服务器公网 | 服务器 -》 电脑显示
用AI Thinker ESP32-CAM板子访问公网ip的5112端口并上传你的摄像头拍摄的图像视频数据,并写一段python程序打开弹窗接受图像实现超远程图像传输教程免费 1. 首先你要有一个公网ip也就是去买一台拥有公网的服务器电脑,我买的是腾讯云1年38元的服务器还可…...

今日踩坑之@Autowired与@Resource区别
案例说明 今天算是体验到了这俩注解的区别了,真所谓不报错就记不住这两注解区别,其实之前本人也是知道这俩注解的区别的只是我们往往项目上用习惯了Autowired就忘了,看来只有真正体会到报错才能记忆犹新o(╥﹏╥)o。 说明一下本人的踩坑情况&…...

在 M1 芯片的 Mac 电脑上安装 Redis 可以通过 Homebrew 快速完成
以下是详细步骤: 1. 安装 Homebrew(若未安装) 打开 终端,执行以下命令安装 Homebrew(已安装可跳过): /bin/bash -c "$(curl -fsSL https://raw.githubusercontent.com/Homebrew/…...

【从零开始学习JVM | 第一篇】快速认识JVM
什么是JVM? JVM--Java虚拟机,它是Java实现平台无关性的基石。 Java程序运行的时候,编译器将Java代码编译为平台无关的Java字节码文件(.class),接下来对应平台的JVM对字节码进行运行解释,翻译成…...

linux 里的创建了一个tomcat用户 怎么禁止该用户使用crontab 功能
在 Linux 系统中,可以通过以下方法禁止 tomcat 用户使用 crontab 功能: 方法 1:通过 /etc/cron.deny 文件限制 这是 最推荐的标准方法,适用于所有主流 Linux 发行版(CentOS、Ubuntu 等)。 操作步骤&#…...
)
Elasticsearch入门指南(二)
一、RestClient操作索引库 什么是RestClient? ES官方提供了各种不同语言的客户端,用来操作ES。这些客户端的本质就是组装DSL语句,通过http请求发送给ES。官方文档地址: https://www.elastic.co/guide/en/elasticsearch/client/in…...

Mac监控新风尚:酷炫界面,性能监控更直观!
你是否曾经希望自己的Mac能够像Windows那样,轻松一按就能查看任务管理器来监控性能状态呢? 对于Mac用户来说,系统性能监控一直是个挑战——苹果公司并没有提供一个直观的性能监控工具,用户往往需要通过复杂的活动监视器来获取这些…...

碰一碰发视频源码开发深度解析,定制化开发
在移动应用开发领域,便捷的数据分享功能始终备受关注。碰一碰发视频这一创新功能,借助近场通信(NFC)技术,为用户提供了一种快速、直观的视频分享体验。本文将深入剖析碰一碰发视频功能的源码开发过程,涵盖从…...

基于 AI智能体、大模型、RAG、Agent 等技术构建公司内部闭环智能问答系统的详细方案,结合 Spring Boot + Vue 管理系统 的改造思路
以下是基于 AI智能体、大模型、RAG、Agent 等技术构建公司内部闭环智能问答系统的详细方案,结合 Spring Boot Vue 管理系统 的改造思路: 1. 系统架构设计 核心组件 数据源层 内部文档库:公司知识库(如操作手册、FAQ、流程文档&a…...

红宝书第三十四讲:零基础学会单元测试框架:Jest、Mocha、QUnit
红宝书第三十四讲:零基础学会单元测试框架:Jest、Mocha、QUnit 资料取自《JavaScript高级程序设计(第5版)》。 查看总目录:红宝书学习大纲 一、单元测试是什么? 就像给代码做“体检”,帮你检查…...

视觉分析AI赋能智慧水务多场景应用
利用视觉分析的方式智慧水务的AI算法方案 一、背景 在传统水务监管中,面临着诸多痛点。例如,对于河道污染、水面异常、河湖四乱问题以及水生态环境差等情况,传统监管效率低下,预警不及时,资源分散,监控功…...

网络2 IP与MAC IP地址
IP地址与MAC地址 一 1.关系: 网卡有唯一的物理地址:MAC地址,IP是配置在网卡上的逻辑地址。 IP地址、MAC地址是进行网络通信必不可少的 2.MAC地址是物理地址,不可更改,通常与IP地址绑定 3.MAC地址:48位 IP地…...

spark-Core
运行架构 核心组件 由上图可以看出,对于 Spark 框架有两个核心组件: Driver,Spark 驱动器节点 其中Spark 驱动器节点,用于执行 Spark 任务中的 main 方法,负责实际代码的执行工作。 Driver 在 Spark 作业执行时主要…...
)
前端开发中的问题排查与定位:HTML、CSS、JavaScript(报错的解决方式)
目录 1.html 1. 结构错误调试:标签未正确嵌套 2. 语法问题调试:缺失引号 3. 断点调试:动态生成内容时的 JavaScript 错误 4. 网络调试:资源加载错误 5. 性能调试:页面加载性能 总结: 2.CSS 1. 定位…...

VMware Fusion Pro 13 for Mac虚拟机
VMware Fusion Pro 13 for Mac虚拟机 文章目录 VMware Fusion Pro 13 for Mac虚拟机一、介绍二、效果下载 一、介绍 VMware Fusion Pro for Mac,是一款mac虚拟机软件,跟Parallels Desktop一样,都可以让你的 Mac 同时运行一个或多个不同的操作…...
、continue(IDEA插件)、cherry-studio玩转MCP)
使用cline(VSCode插件)、continue(IDEA插件)、cherry-studio玩转MCP
安装环境 uv(python) 为什么不用pip? 使用 uv 时无需进行特定安装。使用 uvx 直接运行。 ⚡️ 比pip快10-100x https://github.com/pypa/pip https://ossinsight.io/analyze/pypa/pip?vsastral-sh%2Fuv#overview 安装 https://github…...
Kotlin FragmentTransaction多容器管理多个fragment
在Activity中管理五个Fragment的切换显示和隐藏,并且希望将这部分逻辑进行封装。之前已经教过他们如何在Kotlin中使用FragmentTransaction进行基本的添加、隐藏、显示和替换操作,现在需要进一步封装这些操作,提高代码的可维护性和复用性。 管…...

PyCharm显示主菜单和工具栏
显示主菜单 新版 PyCharm 是不显示主菜单的,要想显示主菜单和工具栏,则通过 “视图” → “外观” ,勾选 “在单独的工具栏中显示主菜单” 和 “工具栏” 即可。 设置工具栏 此时工具栏里并没有什么工具,因此我们需要自定义工具…...

WebView2最低支持.NET frame4.5,win7系统
WebView2最低支持.NET frame什么版本 WebView2 对 .NET Framework 的最低版本要求 基础支持范围 WebView2 官方支持的 .NET Framework 最低版本为 4.5,同时兼容 .NET Core 3.0 及以上版本18。对于 WPF、WinForms 等桌面应用开发,需确…...

ClickOnce 部署
1、在远程服务器172.16.9.252共享文件文件夹Bluetooth. 2、设置版本自动更新. 3、设置部署 4、设置创建桌面菜单 二、远程发布IIS即可...

Kotlin 中 集合 Collection 的扩展方法完全指南
Kotlin 中 Collection 的扩展方法完全指南 “代码是最美的诗篇”——本文将带你进入 Kotlin 集合扩展函数的世界,帮助你写出既高效又优雅的代码 🚀 一、引言 🤔 在 Android 开发中,集合(Collection)的操作…...

STM32F407使用ESP8266连接阿里云并上传数据
文章目录 前言一、ESP01S介绍1.ESP01S2.MQTT固件烧录3.WIFI连接 二、阿里云平台介绍1.创建产品及添加设备2.连接云平台 三、数据上报四、命令下发五、完整工程 前言 在实现OTA功能我们必须学会如何连接云平台,本文会仔细介绍使用STM32F407和ESP8266连接阿里云平台&…...

TorchServe部署模型-index_to_name.json
在TorchServe部署模型时,若要将模型输出结果映射到指定标签(如分类任务的类别名称),需通过index_to_name.json文件定义索引与标签的映射关系,并在打包模型时将其作为额外文件包含。以下是完整流程和命令示例࿱…...

每日一题——BMN3 小红炸砖块
“落下”操作只会对y轴有影响,所以注意y轴的变化即可 只要给出的坐标有砖块,就遍历查找他的上面是否有砖块,每一层都是这样,直到到没有砖块的那一层; 注意:定义矩阵时要注意,给出的坐标都是大…...

AWS服务器 磁盘空间升级到100G后,怎么使其生效?
在AWS(Amazon Web Services)上扩展EBS(Elastic Block Store)卷的大小后,服务器操作系统并不会自动识别新增的空间。要使操作系统识别并使用新增的磁盘空间,您需要进行一些额外的步骤。以下是详细的指导和说…...
概述)
AWS弹性容器服务(AWS Elastic Container Service,ECS)概述
李升伟 编译 标签:AWS | ECS | 容器 | Docker AWS弹性容器服务(AWS Elastic Container Service,ECS)简介 AWS弹性容器服务(ECS)是一项完全托管的容器编排服务,支持运行、管理和扩展容器化应用…...

【消息队列kafka_中间件】一、快速入门分布式消息队列
在当今大数据和分布式系统盛行的时代,消息队列作为一种关键的中间件技术,发挥着举足轻重的作用。其中,Apache Kafka 以其卓越的性能、高可扩展性和强大的功能,成为众多企业构建分布式应用的首选消息队列解决方案。本篇文章将带你深…...

【Axure元件分享】移动端滑动拨盘地区级联选择器
在移动端产品设计中,地区级联选择器(省/市/区)是用户信息录入场景的核心组件,尤其在电商收货地址、政务信息填报等高频业务中直接影响表单转化率。本文将介绍一款基于Axure的三级动态联动机型地区选择器,通过仿真级联滚…...

宁德时代25年校招演绎数字推理SHL测评题库题型及真题分析
非常感谢您对宁德时代的关注。祝贺您通过宁德时代校园招聘的专业面试环节,现邀请您参与完成以下测评。本轮共两份测评,每份测评对您的最终结果都非常重要,请务必在收到测评后48小时内完成!本测评需要进行远程监考,如果您无法或不愿…...

Python-Django+vue宠物服务管理系统功能说明
❥(^_-) 上千个精美定制模板,各类成品Java、Python、PHP、Android毕设项目,欢迎咨询。 ❥(^_-) 程序开发、技术解答、代码讲解、文档,💖文末获取源码+数据库+文档💖 💖软件下载 | 实战案例 💖文章底部二维码,可以联系获取软件下载链接,及项目演示视频。 本项目…...
)
洛谷普及B3691 [语言月赛202212] 狠狠地切割(Easy Version)
题目:[语言月赛202212] 狠狠地切割(Easy Version) 题号:B3691 难度:普及一 末尾包含对二分法优化的详细解释 题目分析 最后一句应该是本题的考查关键,关于筛选算法的时间优化, 但从功能理论上,我找到了…...
)
FPGA_BD Block Design学习(一)
PS端开发流程详细步骤 1.第一步:打开Vivado软件,创建或打开一个工程。 2.第二步:在Block Design中添加arm核心,并将其配置为IP核。 3.第三步:配置arm核心的外设信息,如DDR接口、时钟频率、UART接口等。 …...

Collection vs Collections:核心区别与面试指南
Collection vs Collections:核心区别与面试指南 一、本质区别(核心记忆点) 维度CollectionCollections身份集合框架的根接口操作集合的工具类包位置java.utiljava.util是否可实例化❌ 接口✅ 类(但构造器私有,不可实…...

sqlite3基本语句
创建表 CREATE TABLE student ( id INTEGER PRIMARY KEY, -- 学号,主键 name TEXT NOT NULL, -- 姓名,不能为空 age INTEGER, -- 年龄 gender TEXT -- 性别 ); SQLite常用数据类型 主键 …...

jupyter notebook 显示conda虚拟环境
使用 nb_conda_kernels 安装 nb_conda_kernels:这个包可以自动从你的 Conda 环境中发现并列出内核。 conda activate base # 确保你在 base 环境或任何其他环境中安装 conda install nb_conda_kernels显示jupyternotebook当前所在的位置。...
岗位笔试机考题)
华为海思IC前端中后端(COTXPU)岗位笔试机考题
近期华为海思即将开始IC设计实现实习岗位机考。小编今天给大家分享下华为海思相关机考题目。 华为海思2025届校招笔试面试经验分享 每年IC秋招笔试其实也是从题库中随机抽出一些题。 华为海思2025届校招笔试面试经验分享华为海思机考主要分三个方向,分别是物理方向…...

HarmonyOS NEXT 实现滑动拼图验证码功能
大家好,我是 V 哥。 在 Gitee 上看到一个用 Java 实现的 HarmonyOS 滑动拼图验证码功能,已经太老了,鸿蒙开发推荐使用 ArkTS 语言,V 哥来改造一下。 以下是基于 ArkTS 的实现方案。由于鸿蒙系统的特性差异,这里提供核…...